Computational Approach for Protein Structure Prediction
Article information
Abstract
Objectives
To predict the structure of protein, which dictates the function it performs, a newly designed algorithm is developed which blends the concept of self-organization and the genetic algorithm.
Methods
Among many other approaches, genetic algorithm is found to be a promising cooperative computational method to solve protein structure prediction in a reasonable time. To automate the right choice of parameter values the influence of self-organization is adopted to design a new genetic operator to optimize the process of prediction. Torsion angles, the local structural parameters which define the backbone of protein are considered to encode the chromosome that enhances the quality of the confirmation. Newly designed self-configured genetic operators are used to develop self-organizing genetic algorithm to facilitate the accurate structure prediction.
Results
Peptides are used to gauge the validity of the proposed algorithm. As a result, the structure predicted shows clear improvements in the root mean square deviation on overlapping the native indicates the overall performance of the algorithm. In addition, the Ramachandran plot results implies that the conformations of phi-psi angles in the predicted structure are better as compared to native and also free from steric hindrances.
Conclusions
The proposed algorithm is promising which contributes to the prediction of a native-like structure by eliminating the time constraint and effort demand. In addition, the energy of the predicted structure is minimized to a greater extent, which proves the stability of protein.
I. Introduction
Resolving the functional conformation of a protein remains a central challenge in molecular biology [1]. Structural knowledge is imperative to analyze the functionality of protein; such knowledge contributes much to the development of better drug design, higher-yield crops, and even synthetic bio-fuels. Predicting the three-dimensional (3D) structure of a protein is one of the greatest challenge and a cornerstone for structural biologists.
Currently, there are three experimental techniques, namely, X-ray crystallography, nuclear magnetic resonance (NMR) spectroscopy, and electron microscopy, that can be used to determine the 3D structure of protein with accuracy. Due to technical difficulties, the gap between the number of known sequences and the predicted structure is widening, which is a formidable challenge in terms of effort demand and time consumption. Thus, there is a greater need than ever before for a reliable computational method to address the problem of protein structure prediction (PSP) directly from the sequence.
The existing computational methods are categorized into three approaches based on the information used to model the protein. Homology modeling and threading utilize the structural information of similar proteins, whereas the ab initio method does not. The template free approach is followed by ab initio and it has been proven to be better in several cases.
Many successive computational studies have proposed solution for the conformation structure of protein, such as the evolutionary algorithm [2,3], Monte Carlo [4,5], and HP models [6,7]; however, such solutions are not powerful to search structures in a huge conformational space. The genetic algorithm (GA) has been chosen and has proven to be a reliable computational tool to address the PSP problem because of 1) its success in solving a complex problem by improving the speed using parallelization and 2) the computation process involved in the genetic search to find the optimal strategies for a large space.
Even though several studies have explored the implementation of GA variants [1,8-17] on PSP, the efficiency of GA can be realized only by setting appropriate parameter values. As various inputs require different parameter settings, they are usually obtained by ad-hoc experimentation with manual tuning by a trial-and-error method. This is time consuming and requires a lot of experience and knowledge. On the other hand, inappropriate selection of values leads to premature convergence and consequently, failure to find the optimum in a reasonable computational time, especially when the size of the conformational space is huge (PSP).
From the research work [18] using GA, it is clear that the confirmation results produced were not satisfactory. To overcome the previously mentioned limitations, the most important parameter values of genetic operators need to be self-organized. Therefore, this study focuses on the application of a novel genetic algorithm called the self-organizing genetic algorithm for PSP (SOGA-PSP), which self-configures the crossover and mutation rate during execution, thereby reducing the complexity, which in turn increases the performance efficiency.
Since Met-enkephalin (Protein Data Bank [PDB] ID: 1PLW) has been a test case in many studies [16,19,20], the developed algorithm was tested using the same protein. In addition, the NNFGAIL segment from islet amyloid polypeptide (PDB ID: 3DGJ) was also tested.
II. Methods
1. Genetic Algorithm
Algorithm begins with initial population
Fitness function f(x) is calculated for every chromosome x in the population.
-
Reiterate the following procedure to regenerate the population;
Choose the parents on the basis of fitness score
Reproduce the offspring by crossover operator
Mutate the resultant chromosome
Calculate the fitness score for elite selection
Resultant chromosome reproduces the population
Algorithm terminates as it reaches the fitness value/number of generations.
2. Self-Organizing Genetic Algorithm
A self-organizing system operates based on information received locally without the guidance of the external environment. It is an adaptive methodology adopted for GA to discover the interaction of operators to collect the required information to set the parameter values in an automated way. The SOGA was developed with full knowledge of the test problem and parameter values which are incremented uniformly in each generation. Self-organization contributes desirable factors to GA by adding knowledge to guide and assist the choice of right parameter values.
3. Self-Organizing Genetic Algorithm to Protein Structure Prediction
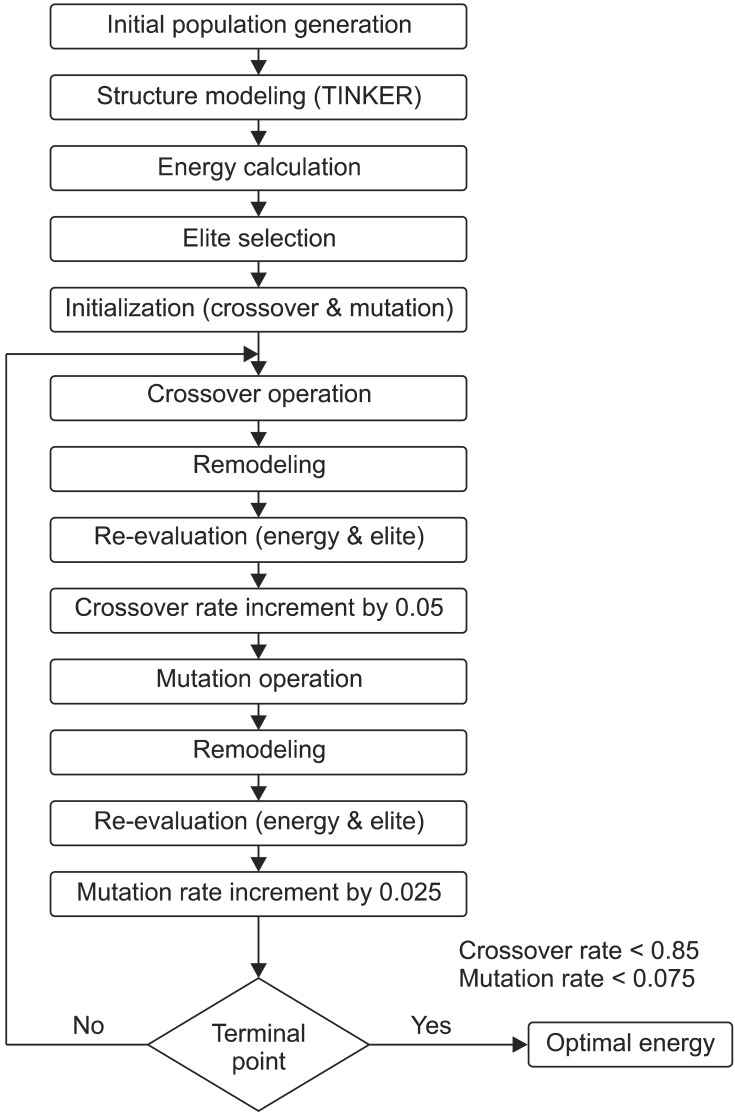
Generally, knowledge regarding the interaction of parameters leads to better global solutions in less time, thereby making GA more robust. A lack of robustness in the choice of design parameters always leads to search imbalance, causing premature convergence and lower performance, which is completely eliminated by the developed algorithm given below:
Step 1: Generate n chromosomes for the initial population
Step 2: Encode the structure for n chromosomes using TINKER [18]
Step 3: Calculate the energy value for each chromosome using Discovery Studio [21]
Step 4: Select and save the elite (chromosome with minimal energy value)
Step 5: Initialize the crossover and mutation rate
-
Step 6: Repeat the following steps to create a new population
Calculate the crossover point for initial crossover
Select the elite and replace, if it is low
Increase crossover rate with uniform interval
Perform mutation with initial mutation rate
Select the elite and replace if it is low
Increase mutation rate by uniform interval
Step 7: Algorithm terminates by reaching the optimal rate SOGA-PSP is pictorially represented in Figure 1.
4. Chromosome Representation
The chromosome representation varies for different problems, and as a set, it constitutes an individual structure called a gene, a set of solution representatives. In general, Cartesian coordinates, templates, torsion angles, side chains, and simplified residues are considered for chromosome representation in PSP [8]. In the present work, torsion [22] and side chain angles [23] were assumed to be best for encoding the chromosomes since both contribute much to the consistency and confirmation of protein structure.
5. Number of Generations
Self-organizing mutation and crossover rates are uniformly increased until they reach the optimal values; in turn the termination condition decides the number of generations.
6. Population Initialization
Population is created using frequently occurring torsion angles [15] which have the degree of freedom for adequate adaptability of GA; it helps in determining the 3D conformation, which is chosen for population size.
7. Fitness Evaluation
The population created in each generation is evaluated by fitness function for selection of the elite [24]. It projects the nature of each chromosome, and the energy value that determines the elite is calculated using the CHARMM force field chosen as the objective function:
Etot: Total energy (fitness)
Ebs: Energy of stretching bond
Eab: Energy of bending angle
EUB: Energy of Urey-Bradley
Eit: Energy of improper dihedrals
Eta: Energy of torsion angle
EVdW: Energy of van der Waals
Ecc: Energy of charge-charge [18]
8. Selection
The selection operator elitism, selects the best chromosome with minimal energy as elite. In each generation the elite may be replaced by the low-energy chromosome for genetic operations.
9. Self-Organizing Crossover Operator (SOCO)
To refine the search space, the crossover operator which is normally fixed in standard genetic algorithm (SGA) is self-organized. The least optimal rate is initially chosen for execution without termination at the convergence point, and it is increased uniformly until it reaches the optimal upper limit [25], or the rates are modified from high to low [26]. The reported range of the rate (0.70 to 0.85) is considered for self-organization.
The working principle of SOCO
Select crossover point with the initial rate
Perform single-point crossover operation
Genes from two parents within the crossover point are interchanged to reproduce children
Model the protein structure using TINKER [27]
Calculate the energy value to select the elite
Repeat the process with uniform interval
The crossover operation is pictorially represented in Figure 2.

Self-organizing crossover operation.
10. Self-Organizing Mutation Operator (SOMO)
The purpose of mutation is to transform the chromosome to change the conformation and to prevent the population from stagnating at any local optimum. With a fixed mutation rate, the search space is compressed, which leads to premature convergence, and it may not be possible to predict the optimal solution. Self-organization is used to avoid this, and the mutation rate is self-configured. Only the constrained values are considered for the test problem since slight changes may occur in the sequential order of amino acids, which in turn may change the entire structure [17].
The working principle of the SOMO operator is:
Select mutation point with initial rate
Mutate the gene value
Model the protein structure using TINKER
Calculate the energy value to select the elite
Repeat the process with uniform interval
The mutation operation is pictorially represented in Figure 3.
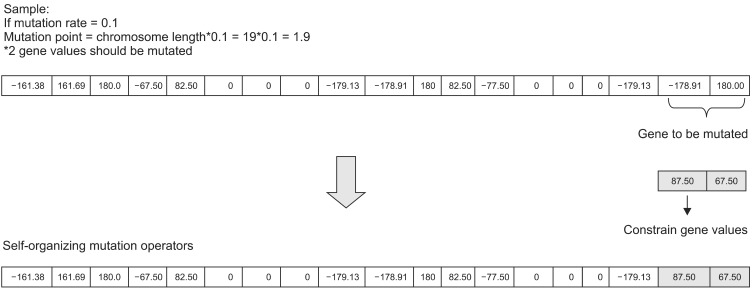
Self-organizing mutation operation.
III. Results
The efficiency of the developed algorithm is implemented on a biological problem, PSP, using two peptides as test case for Met-enkephalin and NNFGAIL.
1. Met-enkephalin
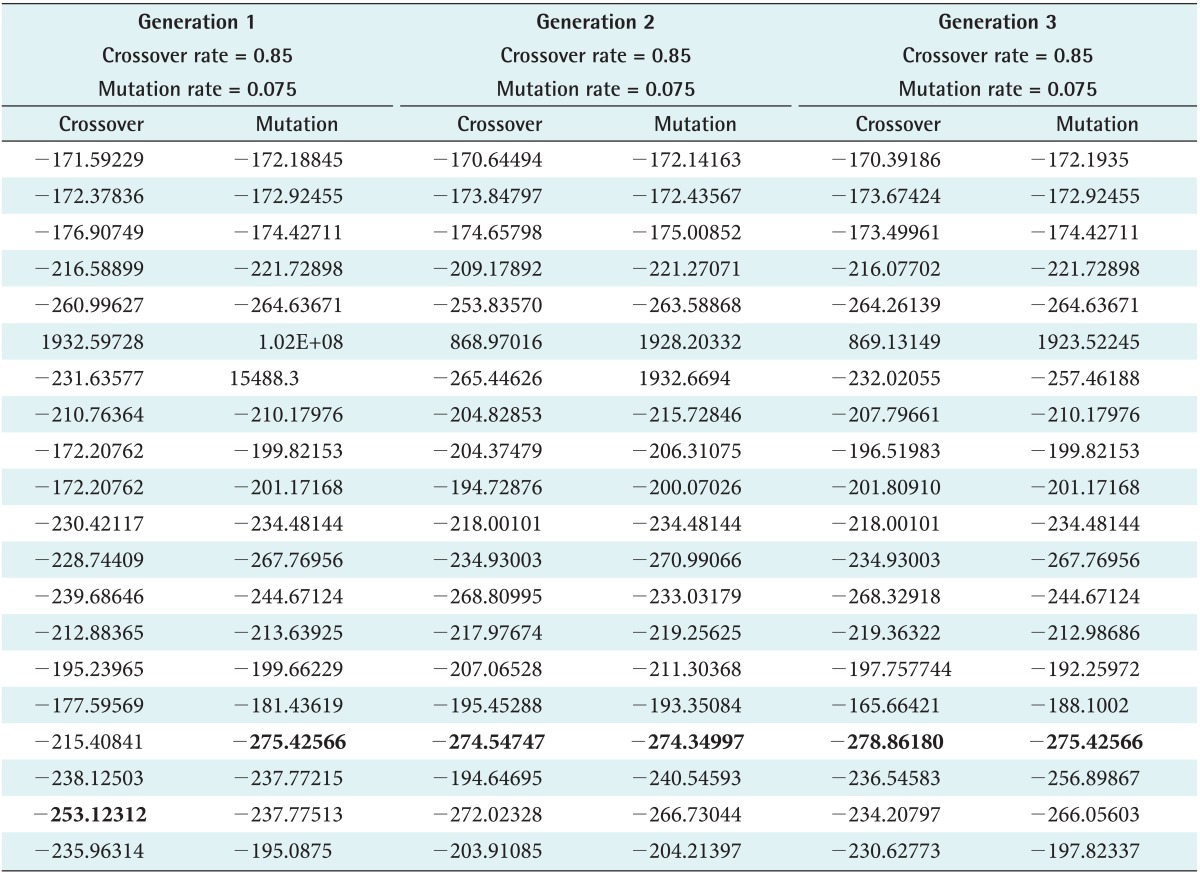
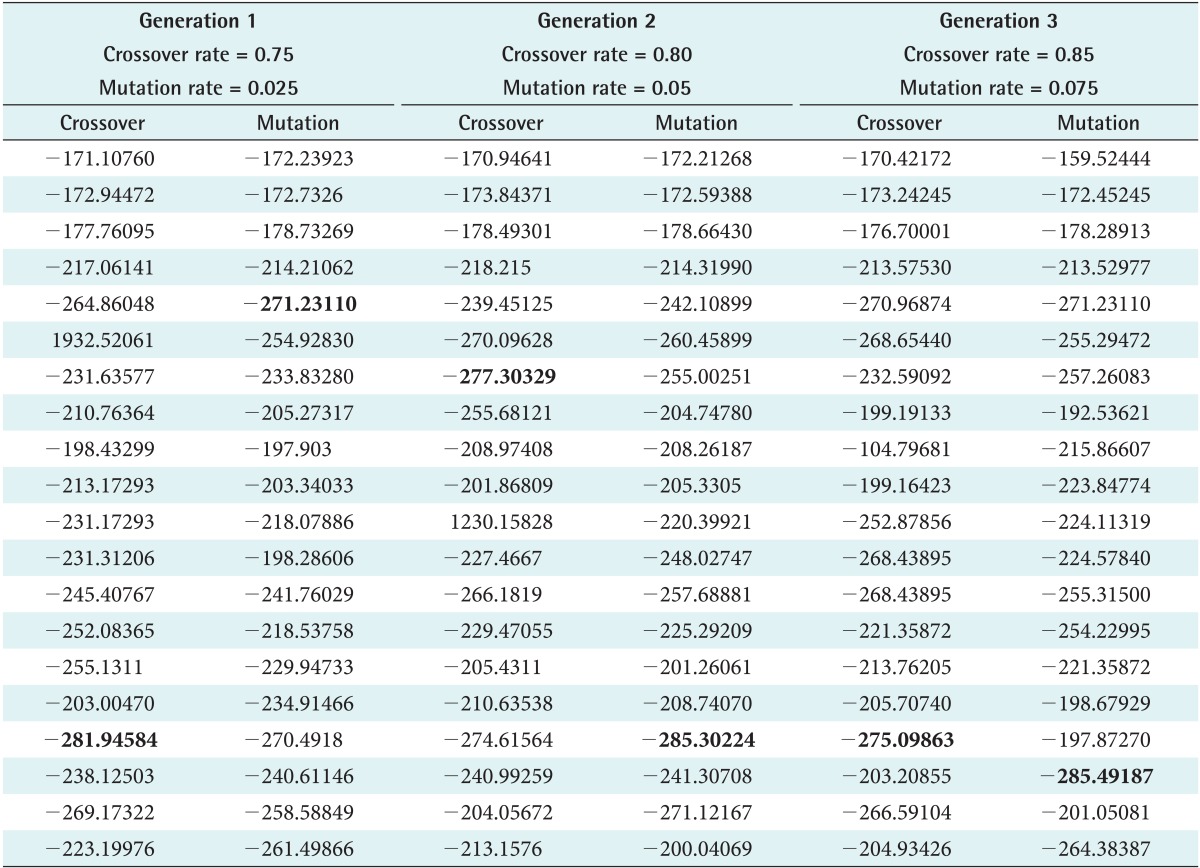
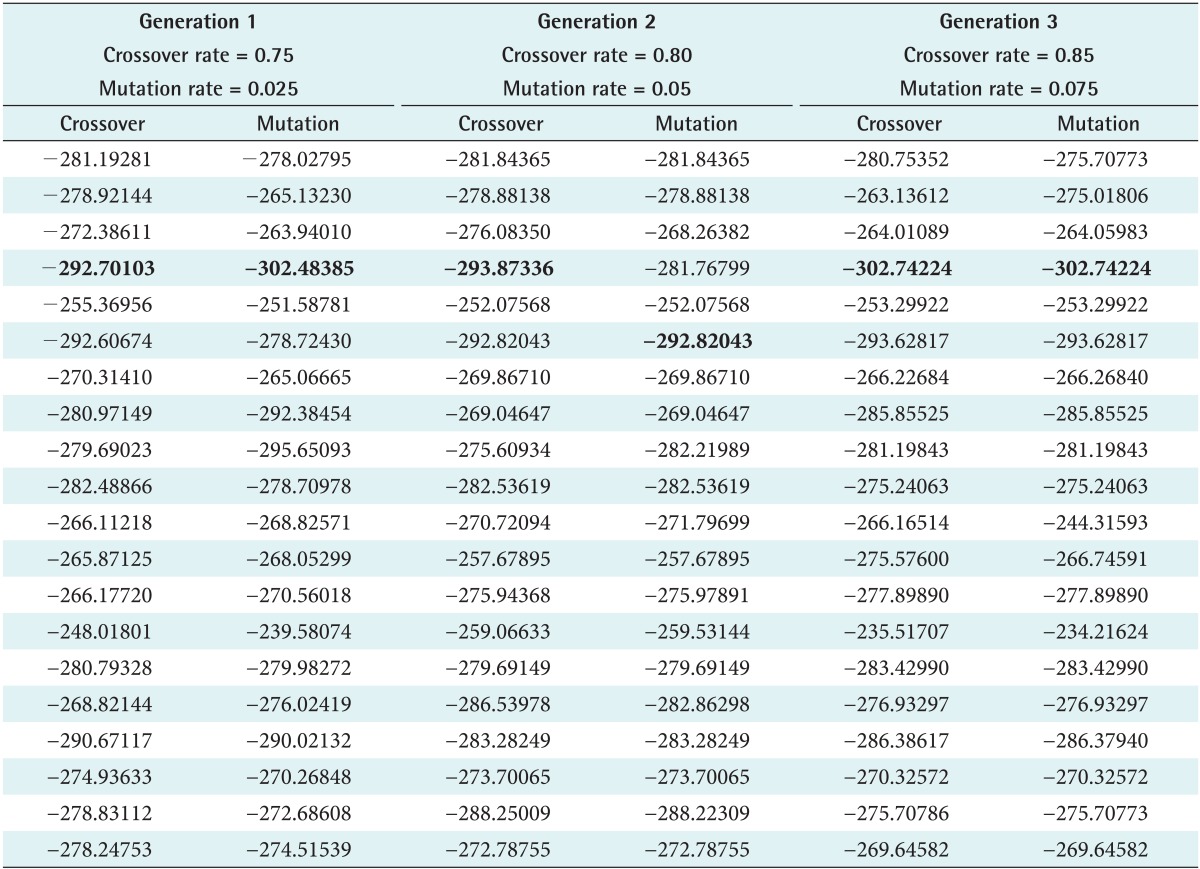
Met-enkephalin (PDB ID: 1PLW) was chosen because it plays a crucial role in generating tissue damage signals as initiated by both peripheral and central nervous system (CNS). Also, it is a simple pentamer used as a test case to measure the efficiency of various ab initio methods. Unlike natural crossover and mutation observed in real cellular chromosomes, randomness is restricted in SGA, where it is designed to propagate in a defined architecture due to fixed values. This limitation is relaxed by defining the rate range so that better results can be produced by switching between the intervals. The rate ranges of 0.75-0.85 and 0.025-0.075 were adopted for crossover and mutation to execute the developed SOGA, whereas the rates are 0.85 and 0.075 in SGA. Accordingly, three generations were produced using predefined rates as shown in Tables 1 and 2. With the inherent limitations of SGA in choosing a crossover rate, peptide conformations are generated, and a huge deviation of energy is observed as seen in the plot. However, in the SOGA approach, predicted conformations are maintained in a standard low-energy distribution as shown in Figures 4 and 5. The best values scored by the protein structure indicate that it provides much better confirmation against a native-structure using NMR. Also similar backbone confirmation is indicated by a root mean square deviation (RMSD) value of 0.0009. Eventually, differences in side chain orientations are also clearly seen; this improves the backbone dihedral angles, which is very clear from Ramachandran plot shown in Table 3 and Figure 6 [28]. On closer view, differences in the orientation of side chains (Y1, F3, and M5) of the predicted and native structures clearly indicate better side chain selection. The overall results confirm the performance efficiency of SOGA-PSP to predict refined peptide structures free from steric hindrances.
1PLW - elite selection with minimal energy value for SGA
1PLW - elite selection with minimal energy value for SOGA (highlighted)

(A) 1PLW - graphical schema of energy variations of crossover in standard genetic algorithm (SGA). (B) Graphical schema of energy variations of mutation in SGA.
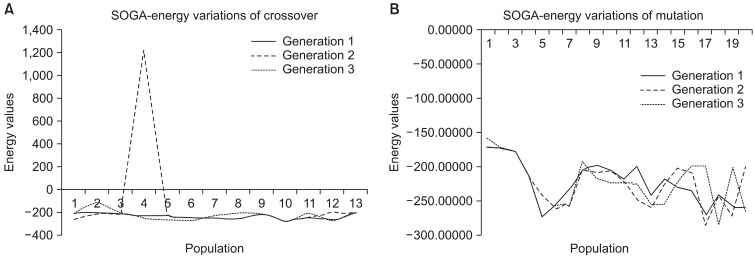
(A) 1PLW - graphical schema of energy variations in crossover of self-organizing genetic algorithm (SOGA). (B) Graphical schema of energy variations in mutation of SOGA.

Ramachandran plot of predicted and native structures of 1PLW

(A) Native structure and its Ramachandran plot of 1 PLW, (B) Predicted structure and its Ramachandran plot. (C) Differences between the native and predicted structures on superimposition.
2. NNFGAIL Segment from Islet Amyloid Polypeptide (3DGJ)
NNFGAIL is a fragment of human islet amyloid polypeptide (IAPP or amylin) of 37-residue hormone found in pancreatic extracts of type 2 diabetics. It plays a role in the function of the pancreas and contributes to glycemic control [29]. The structure of the peptide was predicted using SOGA; as a result, the energy of the structure was comparatively minimal which indicates better conformation as shown in Table 4 and Figure 7. On superimposing with the native structure, an RMSD value of 0.939 was obtained and 5 hydrogen bonds were formed, a most important factor for structure stability. Furthermore, as an advantage out of two hydrogen bonds of X-ray structure, one is conserved. This conservation is also seen in the Ramachandran plot (Table 5 and Figure 8).On further examination, the differences in the orientation of the side chains of the predicted and native structures indicate that better side chain selection is achieved with the proposed approach.
3DGJ - elite selection with minimal energy value for SOGA (highlighted)

(A) 3DGJ - graphical schema of energy variations in crossover of self-organizing genetic algorithm (SOGA). (B) Graphical schema of energy variations in mutation of SOGA.

Ramachandran plot of predicted and native structures of 3DGJ

(A) Native structure and Ramachandran plot of 3DGJ. (B) Predicted structure and its Ramachandran plot. (C) Differences between the native and predicted structures on superimposition.
IV. Discussion
It is well known that the prediction of protein structures through wet lab or experimental methods is very expensive and time consuming. Hence, the challenge was taken up in this study to develop a novel algorithm, SOGA implemented on PSP. The proposed algorithm was proved to be better, and the result obtained demonstrated that it outperforms the approaches taken in previous work [18]. It also avoids the previously mentioned drawbacks of other approaches, which proves the relative efficiency of the algorithm. Furthermore, energy is minimized to a greater extent, and the RMSD value reflects a similar backbone conformation against native structures. The strength of this research also lies in considering only torsion angles as input. In general, the best mechanism adopted for PSP contributes much to the modeling of loops, which are the most common and functionally important substructures. Using current methods like homology modeling or database search, the loops are modeled with less accuracy. As SOGA-PSP has been shown to be a better method to predict the structure of proteins, and it can also be extended to loop modeling.
Acknowledgments
Dr. V. Amouda acknowledges the receipt of financial support from University Grants Commission (UGC), Government of India, and Centre for Bioinformatics, Pondicherry University, India. The authors are grateful to Mr. M. Sundara Mohan and Mr. P. Manivel, Centre for Bioinformatics, Pondicherry University for their constant support all through the work.
Notes
No potential conflict of interest relevant to this article was reported.